Résumé
L’étude des caractéristiques cliniques, épidémiologiques, histologiques et physiopathologiques de la prééclampsie permet de distinguer la prééclampsie précoce, d’origine immunologique, associée à un défaut d’invasion trophoblastique et un retard de croissance intra-utérin, de la prééclampsie tardive d’origine métabolique. Les concepts immunologiques du « soi et du non-soi » et du « manque de soi » utilisés dans l‘immunologie de la défense ne sont pas applicables à l’immunologie de la reproduction. Des processus d’immunotolérance et d’interaction immunologiques entre les cellules utérines maternelles et les cellules trophoblastiques interviennent en effet également dans l’immunologie de la grossesse et de la prééclampsie précoce. Les principaux mécanismes immunologiques de protection du fœtus vis-à-vis de la mère au cours de la grossesse normale sont l’existence d’une diminution de l’expression des antigènes du complexe majeur d’histocompatibilité CMH de classe I et II, la présence de protéines de régulation du complément, la production locale d’IDO, la présence de cellules T régulatrices et de protéines jouant le rôle de points de contrôle immunitaires inhibiteurs. Ces mécanismes de protection sont plus proches de ceux observés au cours d’une prolifération tumorale vascularisée que de ceux observés au cours d’une allogreffe. Le rôle des cellules immunitaires et non immunitaires de la muqueuse utérine maternelle est essentiel dans la régulation de l’invasion du trophoblaste extravilleux et par voie de conséquence dans la régulation de la croissance fœtale et le risque de survenue d’une prééclampsie précoce. Les cellules T régulatrices situées à l’interface fœto-maternelle contribuent à la création d’une immunotolérance locale. Les cellules NK utérines ont, contrairement aux cellules NK circulantes, un phénotype cytokinique et non cytotoxique. Ce phénotype cytokinique est favorisé par la présence de HLA-G et HLA-E et contribue au remodelage vasculaire placentaire quand il est activé. L’activation ou l’inhibition des cellules NK utérines dépend des différentes combinaisons possibles entre les récepteurs KIR des cellules NK et les antigènes HLA-C exprimés par le trophoblaste. L’étude de l’évolution des espèces chez les mammifères montre que la présence de cellules NK, de HLA-G, de récepteurs KIR et de HLA-C1 et C2 est indispensable à la formation d’un placenta hémochorial avec remodelage vasculaire. Ce phénomène placentaire n’est en effet observé que chez les grands singes et l’Homme. Les macrophages et leur phénotype M2 contribuent également à la formation d’une immunotolérance locale et au remodelage vasculaire. Une décidualisation satisfaisante des cellules stromales est également indispensable pour obtenir un remodelage vasculaire du placenta de qualité. La connaissance des mécanismes immunologiques nécessaires à la réalisation d’un remodelage vasculaire adéquat ouvre de nouvelles voies de recherche permettant d’identifier des marqueurs biologiques ou des cibles thérapeutiques susceptibles de mieux prédire ou prévenir la survenue d’une prééclampsie précoce.
1. Service de néphrologie et de dialyse clinique de l’Estrée, 35 rue d’Amiens 93240 Stains
2. Laboratoire d’immunologie de la transplantation, Institut de recherche Saint-Louis, Hôpital Saint-Louis 1, avenue Claude-Vellefaux 75010 Paris
3. Service de gynécologie et d’obstétrique clinique de l’Estrée, 35 rue d’Amiens 93240 Stains
4. UF de fœtopathologie Inserm U1141 Hôpital Robert-Debré, 48 bd Sérurier 75019 Paris
INTRODUCTION
La grossesse ne suscite aucune question particulière quand elle se déroule normalement, ce qui correspond à la majorité des cas. Néanmoins, dans un certain nombre de situations, surviennent des événements indésirables, à type de fausses couches spontanées à répétition, de retard de croissance intra-utérin vasculaire RCIU, d’hématome rétroplacentaire, de mort fœtale in utero, de prééclampsie précoce, d’accouchement prématuré ou d’anémie hémolytique du nouveau-né. L’ensemble de ces affections que l’on regroupe sous le nom de grands syndromes obstétricaux pourrait, en réalité, dans un certain nombre de situations, partager une caractéristique
commune sous-jacente correspondant à celle d’un dysfonctionnement immunitaire entre la mère et l’unité fœto-placentaire. En ce qui concerne le RCIU, l’hématome rétroplacentaire et la prééclampsie précoce, une caractéristique commune sous-jacente supplémentaire correspond à celle d’une placentation défectueuse [1]. Nous aborderons ici uniquement les aspects immunologiques de la prééclampsie précoce et par voie de conséquence ce qui différencie immunologiquement la grossesse normale de la prééclampsie précoce. Dans un premier temps, il est important de planter le décor en précisant clairement ce qui différencie sur le plan clinique, épidémiologique, histologique et physiopathologique la prééclampsie précoce de la prééclampsie tardive et en définissant, à l’aune des connaissances actuelles, le concept immunologique du soi, du non-soi et du manque de soi au cours de la grossesse normale et de la prééclampsie précoce. Dans un deuxième temps, seront décrits les principaux mécanismes immunologiques permettant une protection du fœtus vis-à-vis de la mère. Cette précision préalable permettra de mieux appréhender ce qui différencie sur le plan immunologique une grossesse normale d’une greffe tissulaire et une prééclampsie précoce d’un rejet de greffe tissulaire et, à l’inverse, ce qui rapproche une prolifération tumorale vascularisée d’une grossesse normale. Nous entrerons ensuite dans le cœur du sujet en précisant la participation respective des principaux acteurs immunologiques cellulaires situés à l’interface fœto-maternelle dans la régulation de l’invasion du trophoblaste extravilleux, du remodelage vasculaire placentaire des artères spiralées utérines et par voie de conséquence, dans le retentissement de cette régulation sur la croissance fœtale et le risque de survenue d’une prééclampsie précoce. Le rôle des lymphocytes T régulateurs, des cellules Natural Killer NK utérines, des macrophages et de la décidualisation des cellules stromales dans cette régulation sera ici plus particulièrement développé. Au décours de cette analyse seront esquissées les futures voies de recherche permettant potentiellement d’identifier des marqueurs biologiques ou des cibles thérapeutiques susceptibles de prédire ou prévenir la survenue d’une prééclampsie précoce.
2. Les principales distinctions entre la prééclampsie précoce et tardive
Les prééclampsies se subdivisent, classiquement et schématiquement, même si des zones de chevauchement existent, en prééclampsies précoces ou placentaires apparaissant avant 34 semaines d’aménorrhée SA et en prééclampsies tardives ou maternelles survenant à partir et au-delà de 34 SA. Ces deux types de prééclampsies se distinguent par leur présentation clinique, leur épidémiologie, leur histologie placentaire et leur physiopathologie.
2.1. Une distinction clinique
Les prééclampsies précoces surviennent le plus souvent de manière brutale, chez une femme jeune, primipare, n’ayant généralement pas de pathologie vasculaire sous-jacente. Elles ne représentent que 10 à 20 % de l’ensemble des prééclampsies. Elles sont donc plus rares que les prééclampsies tardives, mais sont en revanche plus graves en termes de retentissement néonatal et de morbimortalité fœto-maternelle en raison de la précocité de leur apparition et de leur association à un RCIU. Les prééclampsies tardives sont à l’inverse beaucoup plus fréquentes. Elles surviennent chez des femmes généralement plus âgées ayant déjà eu d’autres grossesses et ayant également des facteurs de risque vasculaires à type d’hypertension, de diabète et d’obésité. Elles ne s’accompagnent généralement pas d’un RCIU.
2.2. Une distinction épidémiologique
L’hétérogénéité des facteurs de risque de la prééclampsie peut être homogénéisée en les subdivisant en deux sous-groupes, comprenant d’une part les facteurs de risque immunologiques allo-immuns correspondant aux prééclampsies précoces et, d’autre part, les facteurs de risques métaboliques correspondant aux
prééclampsies tardives. Un troisième sous-groupe, distinct, plus restreint, correspondant aux prééclampsies secondaires à une thrombophilie soit acquise, s’intégrant dans le cadre d’une maladie auto-immune, avec la présence d’anticorps antiphospholipides, soit congénitale secondaire à un déficit en antithrombine III, en protéines C, S ou à une mutation du facteur V, peut également être rajouté [2, 3].
Les arguments épidémiologiques en faveur d’une origine immunologique de la prééclampsie précoce [32] sont suggérés par l’existence d’une mémoire génétique, d’une spécificité liée au partenaire et d’une tolérance immunologique aux antigènes paternels présents dans le sperme du conjoint. Ces trois critères sont en effet généralement présents au cours des phénomènes immunologiques.
La mémoire génétique est illustrée par l’augmentation du risque de prééclampsie en cas de prééclampsie préexistante avec un passage d’une valeur de 4,1 % au cours d’une première grossesse à une valeur de 14,7 % au cours de la deuxième grossesse en cas de première grossesse compliquée d’une prééclampsie, et à une valeur de 31,9 % au cours de la troisième grossesse en cas de deux grossesses successives antérieures compliquées d’une prééclampsie [4, 5].
La spécificité immunologique est illustrée par la variation de l’incidence de la prééclampsie et du RCIU en fonction du partenaire. Chez une femme ayant eu de multiples grossesses normales, le changement de partenaire entraine une augmentation du risque ultérieur de prééclampsie de 30 % [6, 7]. À l’inverse chez une femme ayant eu une grossesse avec prééclampsie, le changement de partenaire entraine une réduction de 30 % du risque ultérieur de prééclampsie [7].
La tolérance immunologique est illustrée par la plus grande fréquence de prééclampsie chez une primipare, avec un risque de prééclampsie de 4,1 % au cours de la première grossesse et uniquement de 1,7 % au cours des grossesses ultérieures en cas de première grossesse normale. Elle est également illustrée par la diminution du risque de prééclampsie en cas d’augmentation de la durée de cohabitation sexuelle avant la survenue d’une première conception [5, 8]. Cette diminution du risque de prééclampsie chez les multipares et en cas d’augmentation de la durée de cohabitation sexuelle pourrait s’expliquer par un phénomène de désensibilisation et de tolérance de la muqueuse utérine aux antigènes paternels présents dans le sperme du conjoint. Cette absence de sensibilisation préalable au sperme du conjoint a également été proposée pour expliquer le risque plus important de prééclampsie observé au cours des fécondations in vitro avec don de sperme qui persiste même après ajustement des facteurs confondants grossesses multiples, primiparité, âge maternel avancé [9, 10].
L’incidence de la prééclampsie varie également en fonction des territoires géographiques et des populations étudiées. Elle est notamment deux fois plus importante dans les populations d’origine subsaharienne ayant migré en Europe ou aux États-Unis, que dans les populations d’origine européenne, hispanique ou d’Afrique du Nord, et ceci indépendamment du statut socioéconomique et de l’existence d’une hypertension préexistante [11, 12]. Une transmission génétique maternelle, mais également paternelle est aussi observée au cours des clusters familiaux de prééclampsies [3, 14, 15]. Le risque de prééclampsie est enfin 3 à 4 fois plus important en cas de procréation médicale assistée PMA avec don d’ovocyte hétérologue ou en cas de mère porteuse, probablement en raison de la constitution dans ces situations d’une véritable allogreffe [16, 17].
Les facteurs de risque des prééclampsies précoces, tardives et secondaires sont illustrés dans le tableau 1.
| Tableau 1 – Subdivision des facteurs de risque de la prééclampsie en facteurs de risque maternels ou métaboliques correspondant aux prééclampsies tardives, en facteurs de risque immunologiques correspondant aux prééclampsies précoces et en facteurs de risque thrombotiques autoimmuns ou congénitaux correspondant aux prééclampsies secondaires |
| Hypertension chronique préexistante à la grossesse IMC >30 Diabète Maladie rénale chronique Âge > 40 ans Grossesses gémellaires (multifœtales) |
| Origine ethnique (ancêtres subsahariens) Nulliparité ATCD de prééclampsie précoce, RCIU, HRP, Décès à la naissance Facteur paternel Intervalle entre deux grossesses de plus de 5 ans Procréation médicale assistée (don d’ovocyte auto ou hétérologue) Facteurs génétiques ou immunogénétiques (mère, sœurs, père) |
| Syndrome des anticorps antiphospholipides primaire ou s’intégrant dans un Lupus systémique Déficit en antithrombine III, en protéines C, S, mutation du facteur V |
2.3. Une distinction histologique placentaire
Les lésions histologiques placentaires observées au cours de la prééclampsie précoce se manifestent par un défaut de transformation des artères spiralées utérines et par des lésions « d’athérose aigüe » caractérisées par une paroi artérielle remaniée par de la nécrose fibrinoïde associée à une infiltration de macrophages avec inclusion lipidique, une infiltration périvasculaire de cellules mononuclées et une media musculaire parfois persistante [18]. Le défaut de remodelage des artères spiralées utérines est lié à un défaut d’invasion de leur paroi par les cellules du trophoblaste extravilleux. Les artères spiralées utérines conservent par conséquent leur endothélium vasculaire, leur media musculaire et leurs capacités contractiles. L’absence de dilatation physiologique des artères spiralées utérines s’accompagne d’une hypoperfusion placentaire et le plus souvent d’un RCIU [19, 20]. Les prééclampsies tardives ne sont à l’inverse généralement pas associées à un défaut d’invasion trophoblastique et de remodelage vasculaire [19, 21]. Le remplacement de la paroi par les cellules trophoblastiques et la dilatation physiologique des artères spiralées utérines sont conservés. Il n’y a par conséquent pas d’hypoperfusion placentaire précoce et de RCIU. L’ischémie placentaire est ici tardive, secondaire à un effet mécanique compressif lié à la masse du placenta ou à une vascularisation maternelle moins performante, favorisée par les facteurs de risque vasculaire de la mère.
2.4. Une distinction physiopathologique
La prééclampsie précoce et le RCIU d’origine vasculaire partagent le même dysfonctionnement placentaire initial, défini par un défaut de remodelage vasculaire des artères utérines aboutissant à une diminution de la perfusion du fœtus et à une diminution de son poids de naissance. La prééclampsie précoce diffère du RCIU vasculaire par la survenue d’une libération plus importante de facteurs anti-angiogéniques et peut-être par la libération de facteurs placentaires supplémentaires, de nature différente comme les fragments membranaires de syncytiotrophoblaste dans la circulation maternelle responsables d’un dysfonctionnement vasculaire maternel systémique et de l’apparition d’une hypertension et d’une protéinurie [22].
La prééclampsie tardive et la grossesse normale ne diffèrent l’une de l’autre que par la survenue d’un dysfonctionnement endothélial plus précoce avec l’apparition d’une protéinurie et d’une hypertension avant l’accouchement au cours de la prééclampsie tardive. L’étude de Strevens et al. en 2003 a en effet montré que les biopsies rénales réalisées systématiquement chez les femmes ayant une grossesse normale présentaient dans 50 % de cas des lésions d’endothéliose glomérulaires typiques de la prééclampsie, même si celles-ci n’exprimaient pas encore de protéinurie [23]. L’augmentation des facteurs placentaires anti-angiogéniques se traduit à partir d’un certain seuil par un dysfonctionnement endothélial systémique à l’origine de la survenue des manifestations cliniques de la prééclampsie essentiellement définie par une hypertension et une protéinurie. Au cours de la grossesse normale, on observe également une augmentation des facteurs anti-angiogéniques au cours du temps avec une valeur maximale en fin de grossesse qui n’atteint cependant pas la valeur seuil responsable des manifestations cliniques de la prééclampsie associant HTA et protéinurie, en raison de la survenue de l’accouchement et de la délivrance du placenta avant l’atteinte de ce seuil. Certains auteurs considèrent pour cette raison que si la grossesse se prolongeait jusqu’à 50 SA, la survenue d’une prééclampsie serait présente dans 100 % des cas [24]. La prééclampsie tardive et la grossesse normale ne sont donc finalement que deux aspects cliniques d’un continuum physiopathologique sous-jacent identique, ne se distinguant que par une augmentation des facteurs anti-angiogéniques au-dessus de la valeur seuil survenant avant l’accouchement dans le cas de la prééclampsie tardive et par la survenue de l’accouchement avant l’atteinte de la valeur seuil des concentrations circulantes des facteurs anti-angiogéniques dans le cas de la grossesse normale.
3. Les concepts immunologiques du soi, du non-soi et du manque du soi au cours de la grossesse normale et de la prééclampsie précoce
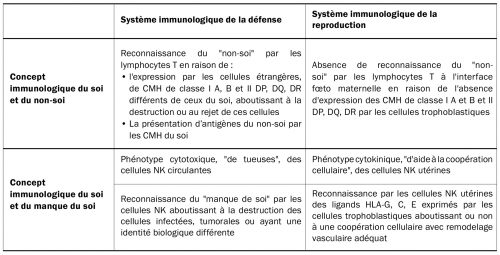
Les organismes biologiques porteurs d’une immunité innée et adaptative se protègent contre la présence d’une cellule, d’un tissu ou d’un organe ayant une identité biologique différente par deux systèmes de reconnaissance, celui du soi et du non-soi, et celui du manque du soi. L’identification du soi et du non-soi est effectuée par les lymphocytes T, qui reconnaissent à la surface de chaque cellule un marqueur du soi qui correspond au complexe majeur d’histocompatibilité CMH. L’identification, par les lymphocytes T, d’une cellule exprimant soit un CMH différent de celui de l’organisme, soit un antigène du non-soi présenté par un CMH du soi, aboutit à la destruction de cette cellule. L’identification du manque du soi est réalisée par les cellules NK et consiste à détruire les cellules qui n’expriment pas de CMH. Ce double système de défense permet de maintenir la cohésion biologique de chaque organisme. Il ne permet pas en revanche de comprendre le développement d’une grossesse, qui correspond à une semi-allogreffe et même à une greffe à part entière en cas de procréation médicalement assistée par don d’ovocyte hétérologue ou avec une mère porteuse. Dans ces deux situations, aucun rejet par le système immunologique de défense de la mère n’est en effet observé. L’immunologie de la défense doit en réalité être distinguée de l’immunologie de la reproduction sur au moins deux points concernant les lymphocytes T et les cellules NK. La reconnaissance tissulaire du trophoblaste et l’activation des lymphocytes T ne peut se réaliser au cours de la grossesse, car le trophoblaste villeux qui est au contact des cellules sanguines maternelles n’exprime aucun antigène du CMH et le trophoblaste extravilleux qui est au contact des cellules de la muqueuse utérine n’exprime pas les antigènes du CMH de classe I A et B et de classe II qui sont les plus immunogènes. Les cellules NK périphériques circulantes, qui représentent un système de défense de l’immunité innée contre les cellules cancéreuses et les cellules infectées qui n’expriment plus le CMH, doivent être par ailleurs distinguées des cellules NK utérines. Les cellules NK utérines impliquées dans l’immunologie de la reproduction n’ont pas, contrairement aux NK circulantes, une fonction cytolytique de « tueuse », mais une fonction cytokinique favorisée par l’immunotolérance locale placentaire. Elles ne sont par ailleurs pas activées par « le manque de soi » au contact des cellules trophoblastiques qui n’expriment pas le CMH de classe I A, B et de classe II. Leur activation est dépendante d’un autre système faisant intervenir l’interaction des récepteurs Killer-cell Immunoglobulin-like Receptors KIR activateurs et inhibiteurs exprimés à leur surface avec les ligands HLA-C, G et E, exprimés par le trophoblaste extravilleux du placenta. L’activation des cellules NK utérines entraine une production de cytokines permettant de stimuler l’angiogenèse, de favoriser l’invasion du trophoblaste et d’obtenir un remodelage vasculaire adéquat des artères spiralées utérines. Certaines combinaisons associant le KIR inhibiteur KIR2DL-1 et le ligand HLA-C2 inhibent les cellules NK et favorisent la survenue d’un retard de croissance intra-utérin RCIU ou d’une prééclampsie précoce, d’autres combinaisons associant le KIR activateur KIR2DS1 et le ligand HLA-C2 permettent à l’inverse l’activation des cellules NK et le développement d’une grossesse normale [25]. Les différences entre l’immunologie de la défense et celle de la reproduction sont représentées dans le tableau 2.
Tableau 2 – Différences des mécanismes de reconnaissance entre le système immunologique de la défense d’un organisme vivant et celui de la reproduction au cours de la grossesse normale et de la prééclampsie précoce
4. Principaux mécanismes d’échappement immunologiques du fœtus vis-à-vis de la mère
4.1. Une diminution de l’expression des antigènes du CMH par le trophoblaste extravilleux et villeux
Comme cela a été dit dans le chapitre précédent, les villosités choriales au contact des cellules du sang maternel n’expriment aucun antigène du CMH et les cellules du trophoblaste extravilleux au contact des cellules de la muqueuse utérine n’expriment que l’antigène polymorphique HLA-C classique de classe Ia et les antigènes peu polymorphiques non classiques de classe 1b HLA-G et E [26-30].
4.2. Une protection contre l’activation du complément par la présence de protéines membranaires de régulation du complément à l’interface fœto-maternelle
Le complément représente l’un des systèmes de défense de l’immunité innée. Son activation peut être à l’origine d’une réaction inflammatoire au niveau du trophoblaste villeux et extravilleux. La présence de protéines régulatrices membranaires situées sur le trophoblaste empêche l’activation du complément et permet d’éviter une inflammation dans les zones où les cellules maternelles sont en contact avec le syncytiotrophoblaste et le trophoblaste extravilleux [31-33]. Le déficit de ces protéines de régulation entraine chez la souris 100 % d’avortement en raison de l’activation du complément et de l’inflammation placentaire qui en résulte [34]. Chez l’Homme, certains avortements spontanés à répétition associés à des dépôts placentaires de C3 pourraient être attribués à un dysfonctionnement de ces protéines régulatrices membranaires [35]. Une étude réalisée en 2006, sur une lignée de souris gestantes, a montré que l’activation du complément placentaire au niveau des cellules trophoblastiques favorisait les fausses couches et les RCIU et s’accompagnait d’une augmentation de la production placentaire de facteur anti-angiogéniques [59]. La recherche par immunohistochimie d’un marquage C5a-9, témoignant d’une activation du complément, sur le placenta des femmes ayant une prééclampsie précoce et tardive par rapport aux grossesses normales appariées pour l’âge gestationnel n’a en revanche jamais été réalisée.
4.3. Une augmentation de l’expression de l’Indoléamine-2,3-dioxygénase IDO à l’interface fœto-maternelle
L’IDO est une enzyme qui catabolise le tryptophane, un acide aminé essentiel, en kynurénine. Elle suscite un intérêt en immunothérapie du cancer, car elle est surexprimée par le micro-environnement d’un certain nombre de tumeurs et participe à l’échappement immunitaire de la tumeur [36-38]. Elle exerce une contre-régulation à l’inflammation en exerçant un effet négatif sur l’activité, la prolifération et la survie des lymphocytes T par deux mécanismes, un premier direct qui consiste à créer une privation, car les lymphocytes T ont besoin du tryptophane pour leur activité et leur survie et un second mécanisme indirect en stimulant l’activité des cellules T régulatrices. La kynurénine exerce également un effet immunomodulateur. L’IDO favorise aussi l’immunotolérance en modifiant le phénotype des cellules présentatrices d’antigène comme les macrophages. Son inhibition pharmacologique ou sa délétion génétique favorise à l’inverse l’immunogénicité. L’IDO est surexprimée au niveau de l’interface fœto-maternelle et joue un rôle dans l’immunotolérance fœto-maternelle et dans le bon déroulement de la placentation [36]. L’inhibition de l’IDO chez la souris entraine un rejet du produit conceptuel allogénique de la souris par les cellules lymphocytaires T [39].
4.4. Une augmentation du nombre de points de contrôle immunitaires inhibiteurs et de cellules T régulatrices à l’interface fœto-maternelle
Le tissu trophoblastique produit, en plus de l’IDO, de nombreuses autres molécules immunosuppressives ciblant les lymphocytes T maternels situés dans la muqueuse utérine et notamment les cytokines apoptotiques Trail et Fas ligand. Le trophoblaste exprime également des points de contrôle immunitaires inhibiteurs comme PD-L1 qui supprime l’activation des lymphocytes T et HLA-G qui bloque l’activité cytolytique des cellules NK [40]. L’expression de CTLA-4 est également augmentée au niveau de l’interface fœto-maternelle et permet de favoriser l’action des cellules T régulatrices [41, 42].
5. Un système de protection immunologique en miroir de celui d’une allogreffe
Au cours d’une grossesse normale, l’absence d’expression d’antigènes immunisants du CMH par le trophoblaste permet d’éviter une infiltration lymphocytaire avec risque de rejet sans diminuer l’immunité maternelle. La production d’anticorps en cas de vaccination pour la grippe ou le Covid-19 est en effet conservée, de même que la production d’anticorps anti-Rhésus et anti-HLA [43]. La muqueuse utérine n’est donc pas infiltrée par des lymphocytes T, mais par des cellules NK utérines qui interagissent avec les antigènes HLA-C, HLA-E et HLA-G du CMH de classe I, exprimés par les cellules du trophoblaste extravilleux. Les proliférations tumorales vascularisées qui peuvent être sur certains points comparées à l’unité fœto-placentaire de la grossesse, qui est également une prolifération cellulaire vascularisée, mais, à la différence du processus tumoral, contrôlée dans le temps et l’espace produisent des mécanismes d’échappement immunologiques similaires contre le système immunitaire de l’hôte avec une diminution de l’expression des antigènes du CMH de classe I et II, une augmentation du nombre de cellules T régulatrices, des points de contrôle immunitaires inhibiteurs et de la production d’IDO [40].
Au cours d’une greffe tissulaire ou d’organe, l’infiltration et le rejet cellulaire effectués par les lymphocytes T, liés à la présence d’antigènes de CMH différents de celui du receveur sont prévenus par la mise en place d’un traitement immunosuppresseur. En l’absence d’une immunosuppression du receveur, les lésions histologiques observées au cours d’un rejet aigu cellulaire de greffe se manifestent par la présence d’une infiltration de cellules mononucléées, essentiellement constituées des lymphocytes T [44].
L’allogreffe représente donc une situation immunologique en
miroir de celle de la grossesse, car dans le premier cas, l’absence de rejet est obtenue par une immunosuppression du receveur alors que dans le second cas, le rejet est prévenu par une diminution de l’expression des antigènes immunogènes du CMH de classe I et II, l’expression locale de points de contrôle immunitaires inhibiteurs et la présence de cellules T régulatrices.
6. Rôle des différentes cellules de la muqueuse utérine dans la régulation de l’invasion du trophoblaste extravilleux et du remodelage vasculaire
Les cellules de la muqueuse utérine situées au contact des cellules du trophoblaste extravilleux sont constituées pour 60 % de cellules stromales non immunitaires et pour 40 % de cellules immunitaires. Ces cellules immunitaires sont elles-mêmes composées pour 10 % de lymphocytes T, pour 20 % de macrophages et pour 70 % de cellules NK utérines [45].
6.1. Les lymphocytes T et les cellules T régulatrices
Les lymphocytes T de la muqueuse utérine sont composés pour 40 % de lymphocyte T CD4 auxiliaires et pour 60 % de lymphocytes CD8 cytotoxiques [46]. Les lymphocytes CD4 de la muqueuse utérine sont eux-mêmes composés d’une proportion particulièrement importante de lymphocytes T régulateurs au cours de la grossesse. Les lymphocytes T régulateurs sont soit centraux d’origine thymique et permettent de prévenir la survenue de maladies auto-immunes, soit sont induits en périphérie par les exo-antigènes, c’est-à-dire les antigènes étrangers provenant du microbiote intestinal ou des antigènes paternels exprimés par les cellules trophoblastiques du fœtus. Le défaut de leur induction entraine pour les antigènes du microbiote une intolérance digestive objectivée par la survenue de maladies inflammatoires chroniques du tube digestif et, chez l’animal, pour les antigènes paternels au cours de la grossesse par la survenue d’une résorption des embryons et d’un défaut du remodelage vasculaire placentaire [47, 48]. L’absence d’induction périphérique des cellules T régulatrices chez les marsupiaux, dont le représentant le plus connu est le kangourou, pourrait expliquer l’impossibilité pour ces animaux de poursuivre la grossesse dans l’utérus et la nécessité d’expulsion du produit conceptuel au stade embryonnaire avec passage dans la poche ventrale pour qu’il puisse poursuivre son développement. L’induction des lymphocytes CD4 en cellules T régulatrices empêche de plus l’activation des autres cellules effectrices CD4 impliquées dans la défense immunitaire et le rejet. La transformation des cellules CD4 en cellules T régulatrices pourrait être favorisée par le micro-environnement immunotolérant local produit par les cellules du trophoblaste. Chez l’animal, les cellules T régulatrices sont induites par les antigènes paternels du fœtus. Cette induction pourrait expliquer l’immunotolérance et la diminution de l’incidence de la prééclampsie chez les femmes multipares ou ayant eu une longue période de cohabitation sexuelle avant la survenue d’une conception. Les femmes qui développent une prééclampsie ont par ailleurs moins de cellules régulatrices dans la muqueuse utérine que les femmes qui développent une grossesse normale [49]. Les lymphocytes CD8 de la muqueuse utérine ont un phénotype moins cytotoxique que les cellules périphériques et présentent un découplage de leur activité cytotoxique. Elles gardent en effet une cytotoxicité dirigée contre les agents infectieux, mais expriment une certaine tolérance vis-à-vis des antigènes fœtaux malgré la présence de CD8 mémoires pour ces antigènes fœtaux [50-52].
6.2. Les cellules Natural Killers utérines
1. Particularités des cellules NK utérines par rapport aux cellules NK circulantes
Les cellules NK circulantes sont, dans 90 % des cas, cytotoxiques et, dans 10 % des cas, cytokiniques. Les cellules NK circulantes cytotoxiques sont des cellules sentinelles spontanément tueuses et immédiatement fonctionnelles, appartenant à l’immunité innée et impliquées dans l’immunologie de la défense. Les cellules NK détruisent selon le mécanisme du soi manquant les cellules qui n’expriment pas le CMH de classe I, c’est-à-dire les cellules infectées par un virus ou une bactérie, les cellules tumorales et les cellules exprimant un CMH de classe I différent comme dans les allogreffes [53]. Ce mécanisme de reconnaissance s’effectue par l’intermédiaire des récepteurs KIR situés sur les cellules NK. Elles ne passent donc pas, comme avec les lymphocytes T et les lymphocytes B, par un processus d’adaptation immunitaire à l’antigène présenté en produisant des récepteurs TCR ou BCR ou des anticorps suite à une recombinaison somatique.
Les cellules NK utérines constituent la proportion la plus importante des cellules immunitaires de la muqueuse utérine. A l’inverse des cellules NK circulantes, elles ont majoritairement un profil cytokinique IFN-γ, GM-CSF, TNF-α, IL-10… favorisé par le micro-environnement local à l’interface fœto-maternelle. Elles sont impliquées dans l’immunologie de la reproduction et plus précisément dans le cycle menstruel, l’implantation de l’embryon, le remodelage vasculaire des artères spiralées utérines maternelles. Le contrôle exercé par les cellules NK utérines sur le caractère invasif et agressif du trophoblaste extravilleux et sur le remodelage vasculaire conditionne le degré de perfusion du fœtus au cours de la grossesse. Une perfusion adéquate entrainera une croissance fœtale normale et un poids de naissance normal. Un défaut de perfusion entrainera à l’inverse un RCIU, avec hypotrophie associée ou non à une prééclampsie précoce.
Les cellules NK fonctionnent par l’intermédiaire des récepteurs activateurs de la famille des Natural Cytotoxicity Receptors NCR qui reconnaissent les molécules de stress exprimées par les cellules cibles, des récepteurs de la famille des lectines de type CD94 NKG2 qui sont soit inhibiteurs CD94 NKG2 A, soit activateurs CD94 NKG2C, NKG2D, des récepteurs de la famille des Leukocyte Immunoglobulin-like Receptors LILR et des récepteurs KIR inhibiteurs ou activateurs très polymorphiques qui reconnaissent les antigènes HLA-A, B, C du CMH de classe I [54]. Les cellules NK utérines, comme cela a été dit auparavant, diffèrent des cellules NK circulantes, car elles ont un phénotype cytokinique et ne sont pas exposées aux antigènes immunisants HLA-A et HLA-B de classe I. Le phénotype cytokinique des cellules NK utérines est favorisé par l’environnement tissulaire local et par l’interaction avec les antigènes HLA-E et HLA-G exprimés par les cellules du trophoblaste. La reconnaissance de l’antigène HLA-G par les récepteurs KIR2DL4 et LILRB1 et de l’antigène HLA-E par le récepteur CD94/NKG2A contribuent à la diminution des propriétés cytotoxiques des cellules NK utérines [55]. L’activation des cellules NK utérines entraine une production d’interféron γ et de facteurs pro-angiogéniques qui favorisent l’invasion trophoblastique et l’angiogenèse placentaire. L’activation ou l’inhibition cytokinique des cellules NK utérines dépend des combinaisons entre les récepteurs KIR et les antigènes polymorphiques HLA-C exprimés par les cellules du trophoblaste extravilleux.
Les cellules NK ont donc une double contrainte, celle de protéger l’individu sur le plan immunologique contre les infections pour qu’il puisse arriver à l’âge de procréer et celle de pérenniser son espèce en permettant sa reproduction par une vascularisation placentaire adéquate.
2. Coévolution des récepteurs KIR des cellules NK et des ligands HLA de classe I dans la phylogénie des différentes espèces et conséquence sur la placentation actuelle chez l’Homme
Parmi les mammifères, on distingue les monotrèmes ou protothériens, les marsupiaux ou métathériens et les mammifères placentaires ou euthériens. Ces derniers peuvent avoir des placentas non déciduaux représentés par les placentas épithéliochoriaux (porcs, chevaux, baleines, lémuriens) ou des placentas déciduaux représentés par les placentas endothéliochoriaux (chiens, chats) et les placentas hémochoriaux (rongeurs, primates) [56, 57]. Parmi les primates, on distingue les prosimiens ou lémuriens de Madagascar, les singes du Nouveau Monde d’Amérique du Sud et de l’Ancien Monde d’Afrique. Les singes de l’Ancien Monde comprennent les grands singes avec les gibbons, les orangs-outans, les gorilles, les chimpanzés et l’Homme. Chez les lémuriens, le trophoblaste ne pénètre pas dans la muqueuse utérine, car il n’y a pas de cellules NK [58]. Chez les singes du Vieux et du Nouveau Monde, l’antigène HLA-E est présent et interagit avec le récepteur non-KIR CD94/NKG2A. L’antigène HLA-G n’est exprimé que chez les singes de l’Ancien Monde, de même que les antigènes HLA-A et HLA-B. HLA-C1 n’est exprimé que chez l’orang-outan. HLA-C1 et HLA-C2 ne sont exprimés que chez les grands singes : le gorille, le chimpanzé et l’Homme. Parallèlement, une maturation des KIR est observée, avec un réarrangement des KIR en haplotype A et B chez l’Homme [54]. Le trophoblaste n’exprime que HLA-C dans l’espèce humaine, qui apparait plus particulièrement impliqué dans l’immunité de la reproduction alors que HLA-A et B apparaissent plus particulièrement impliqués dans l’immunité du rejet et de la défense contre les infections et les processus tumoraux.
3. Conséquences des différentes combinaisons entre les récepteurs KIR des cellules NK utérines et les antigènes HLA-C du fœtus sur le remodelage vasculaire, la croissance fœtale et le risque de survenue d’une prééclampsie précoce
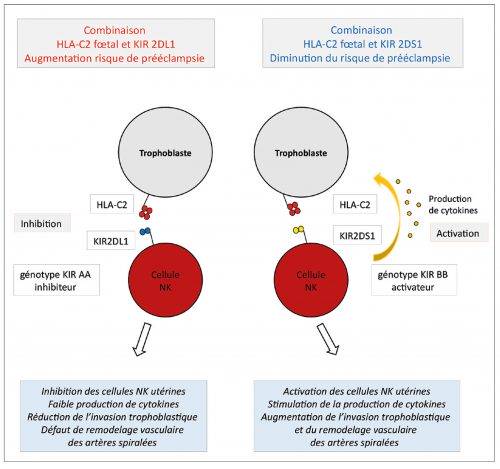
Les récepteurs KIR situés sur les cellules NK utérines sont organisés en haplotypes KIR A et KIR B. Les haplotypes KIR A sont composés de sept gènes qui codent essentiellement pour des KIR inhibiteurs. Les haplotypes KIR B sont composés en moyenne de douze gènes et codent pour des gènes activateurs et inhibiteurs. Les antigènes HLA-C exprimés par les cellules trophoblastiques du fœtus sont subdivisés en deux sous-groupes HLA-C1 et HLA-C2. En 2004 Hiby et al., de l’équipe de Moffett Ashley à Cambridge, ont étudié, sur une population anglo-saxonne, le génotype des récepteurs KIR maternels et des antigènes HLA-C fœtaux dans un groupe de grossesses normales et un groupe de grossesses avec prééclampsie. Ils ont montré que les combinaisons associant l’haplotype homozygote maternel AA avec le couple antigénique fœtal homozygote HLA-C2/HLA-C2 ou hétérozygote HLA-C2/HLA-C1, s’accompagnaient d’une augmentation significative du risque de prééclampsie alors que les combinaisons maternelles KIR BB ou KIR AB avec HLA-C1 ou HLA-C2 se s’accompagnaient pas d’une augmentation du risque de prééclampsie [59]. La combinaison de l’haplotype AA et de HLA-C2 s’accompagne d’une liaison de HLA-C2 au récepteur KIR2DL1 qui est fortement inhibiteur et qui explique pour les auteurs l’inhibition des cellules NK utérines et le défaut de remodelage vasculaire. La combinaison de l’haplotype B avec HLA-C2 s’accompagne à l’inverse d’une liaison de HLA-C2 au récepteur KIR2DS1 fortement activateur et entraine une activation des cellules NK utérines qui favorise l’augmentation du poids de naissance [60]. L’effet activateur de la liaison entre KIR2DS1 et HLA-C2 est supérieur à l’effet inhibiteur de la liaison entre KIR2DL1 et HLA-C2 [32] (Figure 1). Il existe par ailleurs au sein des différentes populations une relation inverse entre la fréquence de l’haplotype AA et la fréquence de HLA-C2. Chez les aborigènes et les Indiens natifs d’Amérique, une prévalence basse de l’haplotype AA avec une prévalence élevée de HLA-C2 est observée. Chez les Japonais, les Coréens et les Chinois, à l’inverse, une augmentation de l’haplotype AA et une baisse de HLA-C2 sont observées, comme s’il existait un système de protection pour éviter le risque trop élevé de survenue d’un RCIU ou d’une prééclampsie et permettre une reproduction et une pérennisation adéquates de l’espèce. Les Européens et les Africains du Nord sont situés entre ces deux extrêmes et ont une fréquence similaire du génotype AA et de HLA-C2 60. Certaines populations comme les populations subsahariennes ne suivent cependant pas ce schéma et ont une fréquence élevée à la fois de l’haplotype AA et de HLA-C2, ce qui pourrait expliquer la plus grande fréquence de complications obstétricales comme le RCIU et la prééclampsie précoce dans cette population [60]. La corrélation robuste mise en évidence entre l’haplotype KIR AA et HLA-C2 et la survenue d’une prééclampsie dans cette étude réalisée en Grande-Bretagne n’a pas été trouvée dans d’autres populations et notamment dans les populations japonaises, ougandaises et danoises [61, 59]. Ces résultats apparemment contradictoires sont probablement liés à des différences alléliques des KIR en fonction des différentes ethnies [61].
Figure 1 – Estimation du risque de prééclampsie précoce en fonction des combinaisons entre les récepteurs KIR utérins et les antigènes HLA-C1 et HLAC-2 fœtaux (adapté de Moffett A et al. Phil. Tans. R. Soc. B 2015)
4. Rôle de HLA-G à l’interface fœto-maternelle
L’antigène HLA-G, qui est exclusivement exprimé par le trophoblaste extravilleux et uniquement chez les mammifères primates, conditionne la placentation en inhibant l’action cytotoxique ou cytolytique des cellules NK et en favorisant leur phénotype cytokinique [62, 63]. Cette action s’effectue par la liaison des différentes isoformes membranaires et solubles de HLA-G aux récepteurs des cellules NK. L’antigène HLA-G se comporte comme un véritable point de contrôle immunologique inhibiteur. Il est en effet également exprimé dans certains cancers où il est considéré comme un facteur de mauvais pronostic, car il inhibe l’activité cytolytique des cellules NK [64]. Des études ont également suggéré que certains variants de HLA-G associés à une diminution de son expression trophoblastique pourraient favoriser la survenue de la prééclampsie [65]. La présence de HLA-G favorise l’expression de HLA-E qui se fixe au récepteur inhibiteur CD94/NKG2A des cellules NK et inhibe leur activité cytolytique. La tolérance induite par HLA-G s’effectue également par un processus de trogocytose, c’est-à-dire par un transfert de membrane de cette protéine d’une cellule à une autre. Ce transfert membranaire de HLA-G a été observé entre les cellules trophoblastiques et les cellules NK utérines, mais également avec les autres cellules déciduales et favorise la création à l’interface fœto-maternelle d’un micro-environnement d’immunotolérance [66]. La fixation des protéines HLA-G membranaires et solubles sur les récepteurs des cellules NK, des cellules T et B et des macrophages conduit à l’inhibition de la fonction de ces cellules et entretient un micro-environnement immunotolérant. Plusieurs études ont par ailleurs montré que les concentrations circulantes de HLA-G solubles, qui pourraient être un marqueur de l’expression membranaire du HLA-G trophoblastique, étaient significativement diminuées au cours de la prééclampsie par rapport à la grossesse normale [67-74]. Aucune étude n’a cependant encore été réalisée pour montrer une possible différence des concentrations circulantes de HLA-G solubles entre les prééclampsies précoces et tardives.
6.3. Les macrophages utérins
Les macrophages représentent, avec 20 % de l’ensemble des cellules immunitaires utérines, la majorité des cellules présentatrices d’antigènes. Au cours de l’implantation, les macrophages inflammatoires de phénotype M1 sont prédominants. Au cours de l’invasion trophoblastique et du remodelage vasculaire, une polarisation vers un phénotype de type M2 apparait progressivement, favorisant l’immunotolérance avec la production d’IDO, d’IL-6, d’IL 10 et de HLA-G, l’homéostasie et la réparation tissulaire. La polarisation M2 devient ensuite prédominante jusqu’à la fin de la grossesse où, de nouveau, un processus inflammatoire est nécessaire pour le déclenchement de l’accouchement [75]. La perturbation de cette polarisation augmente le risque de survenue d’un RCIU, d’une prééclampsie ou d’un accouchement prématuré [76]. La polarisation des macrophages vers M2 est favorisée notamment par la production d’IL-10, de M-CSF et des points de contrôle immunitaires inhibiteurs PD-L1 et HLA-G par le tissu trophoblastique [70].
6.4. Les cellules stromales endométriales
Les cellules stromales endométriales sont initialement des fibroblastes qui se transforment en cellules stromales déciduales suite aux différents stimuli hormonaux survenant au cours du cycle menstruel. Elles représentent 60 % des cellules de la muqueuse utérine. La décidualisation correspond à une modification phénotypique des cellules stromales et à une infiltration de la muqueuse utérine par les cellules immunitaires. Cette maturation endométriale est essentielle à l’implantation du blastocyste, au développement de l’embryon, au remodelage vasculaire des artères spiralées utérines et à la croissance du fœtus. Le défaut de décidualisation s’accompagne d’une augmentation du risque de fausses couches spontanées, de RCIU et de prééclampsies précoces [77-80]. Le défaut de décidualisation peut par conséquent être considéré comme un véritable facteur de risque de prééclampsie précoce. L’étude transcriptionnelle de la déciduale basale en fin de grossesse montre une expression différentielle de 455 gènes pouvant potentiellement être impliqués dans la prééclampsie comparée à la grossesse normale. La ponction trophoblastique réalisée à 11,5 SA montre une expression différentielle de 396 gènes chez les patientes qui développent 6 mois plus tard une prééclampsie par rapport à celles qui développent une grossesse normale. Parmi ces gènes, 112 sont impliqués dans la décidualisation et sont exprimés de manière anormale au cours de la prééclampsie [77]. La production d’annexine A2, un facilitateur de la fibrinolyse, est par exemple diminuée au cours de la décidualisation pendant le cycle menstruel chez la femme prééclamptique et s’accompagne également d’un défaut de décidualisation et d’invasion trophoblastique quand sa production est bloquée chez la souris [81]. L’analyse transcriptionnelle de l’annexine A2 par les cellules déciduales pourrait donc être un exemple de marqueur préconceptuel de la prééclampsie. Les cellules stromales déciduales, par la production de
différentes chémokines, favorisent le recrutement des cellules NK, contribuent au maintien d’un micro-environnement immunotolérant à l’interface fœto-maternelle par la production d’IDO, de points de contrôle immunitaires PD-L1 et HLA-G et de cytokines immunosuppressives TGF-β [82]. L’analyse transcriptionnelle des cellules stromales et de leur décidualisation adéquate au cours du cycle menstruel en période préconceptuelle correspond donc à une nouvelle voie de recherche des facteurs de risque de prééclampsie.
Conclusion et futures voies de recherche
La prééclampsie précoce se distingue sur le plan clinique, épidémiologique, histologique et physiopathologique de la prééclampsie tardive. Les systèmes de reconnaissance et d’interaction cellulaire dans l’immunologie de la défense et du rejet de greffe sont différents de ceux observés dans l’immunologie de la reproduction au cours de la grossesse et de la prééclampsie précoce où d’autres mécanismes de tolérance et d’interactions cellulaires interviennent. Les phénomènes d’échappement immunologiques du fœtus vis-à-vis de la mère associant une diminution de l’expression des antigènes du CMH, la présence de protéines membranaires de régulation du complément, l’augmentation de la production locale d’IDO, de l’expression de points de contrôle immunitaires inhibiteurs tels que PDL1 et HLA-G, et du nombre de lymphocytes T régulateurs à l’interface fœto-maternelle se rapprochent plus des mécanismes immunologiques d’échappement d’une prolifération tumorale cellulaire vascularisée que de ceux d’une greffe où l’absence de rejet est obtenue par une immunosuppression du système immunitaire du receveur. La prééclampsie précoce est donc la conséquence d’un dysfonctionnement de l’interaction immunologique entre les cellules de la muqueuse utérine et les cellules du trophoblaste extravilleux. Un déficit d’induction locale des cellules T régulatrices, une mauvaise combinaison entre les récepteurs KIR des cellules NK utérines et les antigènes polymorphes HLA-C du trophoblaste extravilleux, un défaut de polarisation M2 des macrophages, une décidualisation insuffisante des cellules stromales sont les principaux mécanismes qui peuvent favoriser la survenue d’une prééclampsie précoce. La connaissance de ces différents mécanismes de régulation du remodelage vasculaire placentaire ouvre de nouvelles voies de recherche permettant d’identifier des marqueurs biologiques ou des cibles thérapeutiques pouvant potentiellement prédire ou prévenir la survenue d’une prééclampsie précoce. La détermination préalable en cas de PMA avec don d’ovocyte ou mère porteuse des phénotypes des parents pour éviter une combinaison entre l’haplotype AA et HLA-C2 favorisant la survenue d’une prééclampsie pourrait être réalisée. Les méthodes de thérapie cellulaire avec l’injection de cellules T régulatrices pourraient également être envisagées. L’identification de cibles thérapeutiques permettant d’optimiser les points de contrôle immunitaires inhibiteurs et l’évaluation de la qualité de la décidualisation par l’étude du profil transcriptionnel des cellules déciduales au cours du cycle menstruel des femmes à risque de prééclampsie sont déjà des voies de recherche concrètes pouvant permettre de prédire ou prévenir dans l’avenir la survenue d’une prééclampsie précoce.
Références bibliographiques
[1] Friedman AM, Cleary KL. Prediction and prevention of ischemic placental disease. Semin Perinatol. 2014 Apr;38(3):177-82
[2] Burton GJ, Redman CW, Roberts JM, Moffett A. Pre-eclampsia: pathophysiology and clinical implications. BMJ. 2019 Jul 15; 366:l2381.
[3] Barton JR, Sibai BM. Prediction and prevention of recurrent preeclampsia. Obstet Gynecol. 2008 Aug;112(2 Pt 1):359-72.
[4] Campbell DM, MacGillivray I, Carr-Hill R. Pre-eclampsia in second pregnancy. Br J Obstet Gynaecol. 1985 Feb;92(2):131-40.
[5] Hernández-Díaz S, Toh S, Cnattingius S. Risk of pre-eclampsia in first and subsequent pregnancies: prospective cohort study. BMJ. 2009 Jun 18;338: b2255.
[6] Feeney JG, Scott JS. Pre-eclampsia and changed paternity. Eur J Obstet Gynecol Reprod Biol. 1980 Sep;11(1):35-8.
[7] Li DK, Wi S. Changing paternity and the risk of preeclampsia/eclampsia in the subsequent pregnancy. Am J Epidemiol. 2000 Jan 1;151(1):57-62.
[8] Marti JJ, Herrmann U. Immunogestosis: a new etiologic concept of « essential » EPH gestosis, with special consideration of the primigravid patient; preliminary report of a clinical study. Am J Obstet Gynecol. 1977 Jul 1;128(5):489-93.
[9] Need JA, Bell B, Meffin E, Jones WR. Pre-eclampsia in pregnancies from donor inseminations. J Reprod Immunol. 1983 Nov;5(6):329-38.
[10] Söderström-Anttila V. Pregnancy and child outcome after oocyte donation. Hum Reprod Update. 2001 Jan-Feb;7(1):28-32.
[11] Abalos E, Cuesta C, Grosso AL, Chou D, Say L. Global and regional estimates of preeclampsia and eclampsia: a systematic review. Eur J Obstet Gynecol Reprod Biol. 2013 Sep;170(1):1-7
[12] Abalos E, Cuesta C, Carroli G, Qureshi Z, Widmer M, Vogel JP, Souza JP; WHO Multicountry Survey on Maternal and Newborn Health Research Network. Pre-eclampsia, eclampsia and adverse maternal and perinatal outcomes: a secondary analysis of the World Health Organization Multicountry Survey on Maternal and Newborn Health. BJOG. 2014 Mar;121 Suppl 1:14-24.
[13] Nakimuli A, Chazara O, Byamugisha J, Elliott AM, Kaleebu P, Mirembe F, Moffett A. Pregnancy, parturition and preeclampsia in women of African ancestry. Am J Obstet Gynecol. 2014 Jun;210(6):510-520.e1.
[14] Treloar SA, Cooper DW, Brennecke SP, Grehan MM, Martin NG. An Australian twin study of the genetic basis of preeclampsia and eclampsia. Am J Obstet Gynecol. 2001 Feb;184(3):374-81.
[15] Cnattingius S, Reilly M, Pawitan Y, Lichtenstein P. Maternal and fetal genetic factors account for most of familial aggregation of preeclampsia: a population-based Swedish cohort study. Am J Med Genet A. 2004 Nov 1;130A(4):365-71.
[16] Blázquez A, García D, Rodríguez A, Vassena R, Figueras F, Vernaeve V. Is oocyte donation a risk factor for preeclampsia? A systematic review and meta-analysis. J Assist Reprod Genet. 2016 Jul;33(7):855-63.
[17] Rodriguez-Wallberg KA, Berger AS, Fagerberg A, Olofsson JI, Scherman-Pukk C, Lindqvist PG, Nasiell Increased incidence of obstetric and perinatal complications in pregnancies achieved using donor oocytes and single embryo transfer in young and healthy women. A prospective hospital-based matched cohort study. J.Gynecol Endocrinol. 2019 Apr;35(4):314-319.
[18] Boulanger H, Flamant M. New insights in the pathophysiology of preeclampsia and potential therapeutic implications]. Nephrol Ther. 2007 Dec;3(7):437-48.
[19] Moldenhauer JS, Stanek J, Warshak C, Khoury J, Sibai B. The frequency and severity of placental findings in women with preeclampsia are gestational age dependent. Am J Obstet Gynecol. 2003 Oct;189(4):1173-7. doi: 10.1067/s0002-9378(03)00576-3
[20] Barton JR, Sibai BM. Prediction and prevention of recurrent preeclampsia. Obstet Gynecol. 2008 Aug;112(2 Pt 1):359-72.
[21] Staff AC, Fjeldstad HE, Fosheim IK, Moe K, Turowski G, Johnsen GM, Alnaes-Katjavivi P, Sugulle M. Failure of physiological transformation and spiral artery atherosis: their roles in preeclampsia. Am J Obstet Gynecol. 2020 Sep 21:S0002-9378(20)31116-9
[22] Boulanger H, Drouin D, Largilliere C, Lefèvre G. Intrauterine growth restriction, soluble fms-like tyrosine kinase-1 to placental growth factor ratio increase and preeclampsia. J Gynecol Obstet Hum Reprod. 2019 Oct;48(8):695-697.
[23] Strevens H, Wide-Swensson D, Hansen A, Horn T, Ingemarsson I, Larsen S, Willner J, Olsen S. Glomerular endotheliosis in normal pregnancy and pre-eclampsia. BJOG. 2003 Sep;110(9):831-6. Clinical Trial.
[24] Wright D, Syngelaki A, Akolekar R, Poon LC, Nicolaides KH. Competing risks model in screening for preeclampsia by maternal characteristics and medical history. Am J Obstet Gynecol. 2015 Jul;213(1):62.e1-62.e10.
[25] Moffett A, Loke YW. The immunological paradox of pregnancy: a reappraisal. Placenta. 2004 Jan;25(1):1-8.
[26] Ellis SA, Palmer MS, McMichael AJ. Human trophoblast and the choriocarcinoma cell line BeWo express a truncated HLA Class I molecule. J Immunol. 1990 Jan 15;144(2):731-5.
[27] Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R. A class I antigen, HLA-G, expressed in human trophoblasts. Science. 1990 Apr 13;248(4952):220-3.
[28] Grabowska A, Carter N, Loke YW.Human trophoblast cells in culture express an unusual major histocompatibility complex class I-like antigen. Am J Reprod Immunol. 1990 May;23(1):10-8.
[29] King A, Allan DS, Bowen M, Powis SJ, Joseph S, Verma S, Hiby SE, McMichael AJ, Loke YW, Braud VM. HLA-E is expressed on trophoblast and interacts with CD94/NKG2 receptors on decidual NK cells. Eur J Immunol. 2000 Jun;30(6):1623-31.
[30] King A, Burrows TD, Hiby SE, Bowen JM, Joseph S, Verma S, Lim PB, Gardner L, Le Bouteiller P, Ziegler A, Uchanska-Ziegler B, Loke YW. Surface expression of HLA-C antigen by human extravillous trophoblast. Placenta. 2000 May;21(4):376-87.
[31] Holmes CH, Simpson KL, Wainwright SD, Tate CG, Houlihan JM, Sawyer IH, Rogers IP, Spring FA, Anstee DJ, Tanner MJ. Preferential expression of the complement regulatory protein decay accelerating factor at the fetomaternal interface during human pregnancy. J Immunol. 1990 Apr 15;144(8):3099-105
[32] Morgan BP, Holmes CH. Immunology of reproduction: protecting the placenta. Curr Biol. 2000 May 18;10(10):R381-3.
[33] Holmes CH, Simpson KL, Okada H, Okada N, Wainwright SD, Purcell DF, Houlihan JM. Complement regulatory proteins at the feto-maternal interface during human placental development: distribution of CD59 by comparison with membrane cofactor protein (CD46) and decay accelerating factor (CD55). Eur J Immunol. 1992 Jun;22(6):1579-85.
[34] Xu C, Mao D, Holers VM, Palanca B, Cheng AM, Molina H. A critical role for murine complement regulator crry in fetomaternal tolerance. Science. 2000 Jan 21;287(5452):498-501. doi: 10.1126/science.287.5452.498.
[35] Girardi G, Bulla R, Salmon JE, Tedesco F. The complement system in the pathophysiology of pregnancy. Mol Immunol. 2006 Jan; 43(1-2):68-77. Review.
[36] Chang RQ, Li DJ, Li MQ. The role of indoleamine-2,3-dioxygenase in normal and pathological pregnancies. Am J Reprod Immunol. 2018 Apr;79(4):e12786.
[37] Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int Rev Cell Mol Biol. 2018;336:175-203. doi: 10.1016/bs.ircmb.2017.07.004. Epub 2017 Sep 21. PMID: 29413890 Free PMC article. Review.
[38] Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016 Mar;37(3):193-207.
[39] Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL.Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998 Aug 21;281(5380):1191-3.
[40] Ferreira LMR, Meissner TB, Tilburgs T, Strominger JL. HLA-G: At the Interface of Maternal-Fetal Tolerance. Trends Immunol. 2017 Apr; 38(4):272-286.
[41] Miko E, Meggyes M, Doba K, Barakonyi A, Szereday L. Immune Checkpoint Molecules in Reproductive Immunology. Front Immunol. 2019 Apr 18;10:846.
[42] Zhang YH, Sun HX. Immune checkpoint molecules in pregnancy: Focus on regulatory T cells. Eur J Immunol. 2020 Feb;50(2):160-169.
[43] Colucci F, Kieckbusch J. Maternal uterine natural killer cells nurture fetal growth: in medio stat virtus. Trends Mol Med. 2015 Feb;21(2):60-7.
[44] Solez K, Axelsen RA, Benediktsson H, Burdick JF, Cohen AH, Colvin RB, Croker BP, Droz D, Dunnill MS, Halloran PF, et al. International standardization of criteria for the histologic diagnosis of renal allograft rejection: the Banff working classification of kidney transplant pathology. Kidney Int. 1993 Aug;44(2):411-22.
[45] Bulmer JN, Williams PJ, Lash GE. Immune cells in the placental bed. Int J Dev Biol. 2010;54(2-3):281-94
[46] Yang F, Zheng Q, Jin L. Dynamic Function and Composition Changes of Immune Cells During Normal and Pathological Pregnancy at the Maternal-Fetal Interface. Front Immunol. 2019 Oct 18;10:2317.
[47] Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. 2012 Jul 6;150(1):29-38.
[48] Chavan AR, Griffith OW, Wagner GP. The inflammation paradox in the evolution of mammalian pregnancy: turning a foe into a friend. Curr Opin Genet Dev. 2017 Dec;47:24-32.
[49] Sasaki Y, Darmochwal-Kolarz D, Suzuki D, Sakai M, Ito M, Shima T, Shiozaki A, Rolinski J, Saito S. Proportion of peripheral blood and decidual CD4(+) CD25(bright) regulatory T cells in pre-eclampsia. Clin Exp Immunol. 2007 Jul;149(1):139-45.
[50] Liu L, Huang X, Xu C, Chen C, Zhao W, Li D, Li L, Wang L, Du M. Decidual CD8+T cells exhibit both residency and tolerance signatures modulated by decidual stromal cells. J Transl Med. 2020 Jun 1;18(1):221.
[51] Van der Zwan A, Bi K, Norwitz ER, Crespo ÂC, Claas FHJ, Strominger JL, Tilburgs T. Mixed signature of activation and dysfunction allows human decidual CD8+ T cells to provide both tolerance and immunity. Proc Natl Acad Sci U S A. 2018 Jan 9;115(2):385-390.
[52] Morita K, Tsuda S, Kobayashi E, Hamana H, Tsuda K, Shima T, Nakashima A, Ushijima A, Kishi H, Saito S. Analysis of TCR Repertoire and PD-1 Expression in Decidual and Peripheral CD8+ T Cells Reveals Distinct Immune Mechanisms in Miscarriage and Preeclampsia. Front Immunol. 2020 Jun 3;11:1082.
[53] Malmberg KJ, Carlsten M, Björklund A, Sohlberg E, Bryceson YT, Ljunggren HG. Natural killer cell-mediated immunosurveillance of human cancer. Semin Immunol. 2017 Jun;31:20-29.
[54] Parham P, Norman PJ, Abi-Rached L, Guethlein LA. Human-specific evolution of killer cell immunoglobulin-like receptor recognition of major histocompatibility complex class I molecules. Philos Trans R Soc Lond B Biol Sci. 2012 Mar 19;367(1590):800-11.
[55] Jabrane-Ferrat N. Features of Human Decidual NK Cells in Healthy Pregnancy and During Viral Infection. Front Immunol. 2019 Jun 28;10:1397.
[56] Moffett A, Colucci F. Co-evolution of NK receptors and HLA ligands in humans is driven by reproduction. Immunol Rev. 2015 Sep;267(1):283-97.
[57] Montiel JF, Kaune H, Maliqueo M. Maternal-fetal unit interactions and eutherian neocortical development and evolution. Front Neuroanat. 2013 Jul 19;7:22.
[58] Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol. 2002;20:217-51.
[59] Hiby SE, Walker JJ, O’shaughnessy KM, Redman CW, Carrington M, Trowsdale J, Moffett A.Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med. 2004 Oct 18;200(8):957-65.
[60] Moffett A, Hiby SE, Sharkey AM. The role of the maternal immune system in the regulation of human birthweight. Philos Trans R Soc Lond B Biol Sci. 2015 Mar 5;370(1663):20140071. doi: 10.1098/rstb.2014.0071. PMID: 25602075
[61] Yang X, Yang Y, Yuan Y, Liu L, Meng T. The Roles of Uterine Natural Killer (NK) Cells and KIR/HLA-C Combination in the Development of Preeclampsia: A Systematic Review. Biomed Res Int. 2020 Mar 28;2020:4808072.
[62] Pazmany L, Mandelboim O, Valés-Gómez M, Davis DM, Reyburn HT, Strominger JL. Protection from natural killer cell-mediated lysis by HLA-G expression on target cells. Science. 1996 Nov 1;274(5288):792-5.
[63] Rouas-Freiss N, Gonçalves RM, Menier C, Dausset J, Carosella ED. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci U S A. 1997 Oct 14;94(21):11520-5.
[64] Curigliano G, Criscitiello C, Gelao L, Goldhirsch A.Molecular pathways: human leukocyte antigen G (HLA-G). Clin Cancer Res. 2013 Oct 15;19(20):5564-71.
[65] Quach K, Grover SA, Kenigsberg S, Librach CL. A combination of single nucleotide polymorphisms in the 3’untranslated region of HLA-G is associated with preeclampsia. Hum Immunol. 2014 Dec;75(12):1163-70.
[66] Rouas-Freiss N, Moreau P, LeMaoult J, Papp B, Tronik-Le Roux D, Carosella ED. Role of the HLA-G immune checkpoint molecule in pregnancy. Hum Immunol. 2021 May;82(5):353-361.
[67] Goldman-Wohl DS, Ariel I, Greenfield C, Hochner-Celnikier D, Cross J, Fisher S, Yagel S. Lack of human leukocyte antigen-G expression in extravillous trophoblasts is associated with pre-eclampsia. Mol Hum Reprod. 2000, 6:88-95
[68] Yie SM, Li LH, Li YM, Librach C. HLA-G protein concentrations in maternal serum and placental tissue are decreased in preeclampsia. Am J Obstet Gynecol 2004 191:525-9.
[69] Hackmon R1, Koifman A, Hyodo H, Glickman H, Sheiner E, Geraghty DE. Reduced third-trimester levels of soluble human leukocyte antigen G protein in severe preeclampsia. Am J Obstet Gynecol 2007, 197:255.1-5.
[70] Marchal-Bras-Goncalves R, Rouas Freiss N, Connan F, Chopin J, Dausset ED, Carosella M, Kirszenbaum M, Guillet JG. Transplantation Proceedings 2001, 33: 2355-9.
[71] Rokhafrooz S, Ghadiri A, Ghandil P, Ghafourian M, Hossaini SH, Daraei N, Najafian M, Rouhizadeh A. Association between HLA-G 14bp Gene Polymorphism and Serum sHLA-G Protein Concentrations in Preeclamptic Patients and Normal Pregnant Women. Immunol Invest 2018, 47(6):558-68.
[72] Biyik I. Maternal Serum soluble HLA-G in complicated pregnancies. J Matern Fetal Neonatal Med, 2014; 27(4): 381-4.
[73] Darmochwal-Kolarz D, Kolerz B, Rolinski J, Leszczynska-Gorzelak B, Olesczczuk J. The concentration of soluble HLA-G protein are elevated during mid gestation and decreased in pre-eclampsia. Folia Histochem Cytobiol 2012, 50 (2): 286-91.
[74] He Y, Chen S, Huang H, Chen Q. Association between decreased plasma levels of soluble human leucocyte antigen-G and severe pre-eclampsia. J Perinat Med 2016; 44(3) 283-90
[75] Xu L, Li Y, Sang Y, Li DJ, Du M. Crosstalk Between Trophoblasts and Decidual Immune Cells: The Cornerstone of Maternal-Fetal Immunotolerance. Front Immunol. 2021 Feb 25;12:642392.
[76] Parasar P, Guru N, Nayak NR. Contribution of macrophages to fetomaternal immunological tolerance. Hum Immunol. 2021 May;82(5):325-331.
[77] Garrido-Gómez T, Castillo-Marco N, Cordero T, Simón C. Decidualization resistance in the origin of preeclampsia. Am J Obstet Gynecol. 2020 Sep 29:S0002-9378(20)31130-3.
[78] Rabaglino MB, Post Uiterweer ED, Jeyabalan A, Hogge WA, and Conrad KP. Bioinformatics Approach Reveals Evidence for Impaired Endometrial Maturation Before and During Early Pregnancy in Women Who Developed Preeclampsia. Hypetension. 2015 Feb;65(2):421-9.
[79] Conrad KP, Rabaglino MB, Post Uiterweer ED. Emerging role for dysregulated decidualization in the genesis of preeclampsia. Placenta. 2017 Dec;60:119-129.
[80] Garrido-Gomez T, Dominguez F, Quiñonero A, Diaz-Gimeno P, Kapidzic M, Gormley M, Ona K, Padilla-Iserte P, McMaster M, Genbacev O, Perales A, Fisher SJ, Simón C. Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc Natl Acad Sci U S A. 2017 Oct 3;114(40):E8468-E8477.
[81] Garrido-Gomez T, Quiñonero A, Dominguez F, Rubert L, Perales A, Hajjar KA, Simon C. Preeclampsia: a defect in decidualization is associated with deficiency of Annexin A2.Am J Obstet Gynecol. 2020 Apr;222(4):376.e1-376.e17.
[82] Erkers T, Nava S, Yosef J, Ringdén O, Kaipe H. Decidual stromal cells promote regulatory T cells and suppress alloreactivity in a cell contact-dependent manner. Stem Cells Dev. 2013 Oct 1;22(19):2596-605.
